![]()
![]()
- Files
- Control Panel
- Move & Rotate
- Display
- Rendering
- Tools
- Mutations
- Torsions
- Preferences
- hardware stereo
- electrostatics
- surface
- tips & tricks
Index
![]()
![]()
![]()
![]()
![]()
![]()
![]()
by N.Guex
& T.Schwede
Electrostatic Potential
In this section, we will learn how to compute electrostatic potentials or open potentials computed by other programs.
The electrostatic potential of proteins - caused by charged side chains and bound ions - plays a role e.g in protein folding and stability, enzyme catalysis or specific protein-protein recognition. In general, electrostatic potentials are visualized either as
- Electrostatic potential map
In this case you can change the contouring value of the potential (in kT/e) in real time with the top and bottom arrow cursors (or by opening the EDM window and clicking on the sigma value). Positive potentials are drawn in blue, negative in red, but these colors can be changed by clicking in the color box of the EDM window.
E.g. the charge distribution in bovine catalytic alpha subunit of cAMP-dependent protein Kinase (1YDR) is forming a characteristic anisotropic electric field produding into the surrounding solvent (Displayed as solid p or wireframe (this setting can be altered in the EMAP preferences):
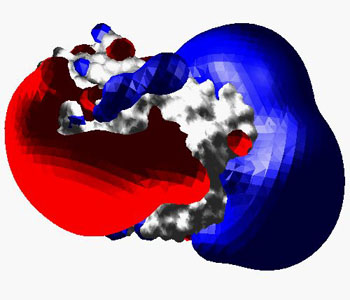
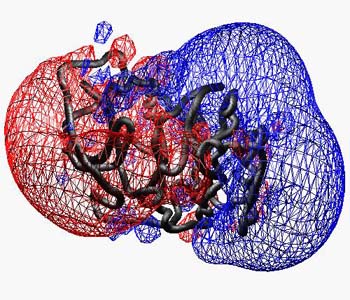
In this image, the ribbon is enabled, and all residues were selected and their secondary structure was assigned as "coil" from the Edit menu in order to get a worm.
- or mapped to a Molecular Surface
To compute a molecular surface with an electrostatic potential, activate the "color by electrostatic potential" in the "Surface" preferences and compute the surface (note that you can also load a surface made by one of the other programs described below). If you activate the "Map potential to surface" option in the "Electrostatics" preferences, new potentials are mapped to an existing surface as they are computed or loaded. This type of surface representations is usefull to discuss e.g. protein interfaces.
The same molecule as above in complex with the peptidic H7 protein kinase inhibitor, but now the potential has been computed at the surface: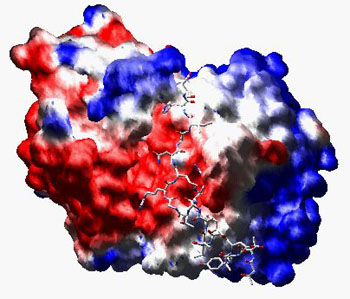
The settings for electrostatic potentials can be altered in the "Electrostatic Potential" Preferences. Currently, the protein is taken to be at pH 7.0, with default protonation state for all residues. As default, only charged residues (Arg, Lsy, Glu, Asp) are taken into account, and the charges are located at the corresponding (non-H) atom positions. You may also use the partial charges of the GROMOS 43A1 force field. This is much slower, as more charged atoms are present.
Normally potentials are computed using simple coulomb interaction. This is the fastest method, but also the less accurate, as only a uniform dielectric constant is applied both for protein interior and for the solvent space. If we want to account for the different dielectric properties of the protein interior and the solvent, we have to solve the the Poisson-Boltzmann equation numerically. (This procedure in not yet implemented in SPDV, but you may display a PB-potential computed with DELPHI).
SPDBV also allows you to visualize surfaces or potentials computed with other programs:
- DELPHI - potential
maps
The program DELPHI computes a numericals solution of the PB-equation. [Honig and Nicholls, Science (1995) 268, 1144-1149] - GRASP - surfaces
GRASP computes molecular surfaces and a simpler numerical PB-solution on a reduced grid. SPDBV reads plain surface files (fort.25, *.srf) with or without electrostaic potentials [Ref: Anthony Nicholls, Kim Sharp and Barry Honig; Proteins (1991) vol. 11 pp 281].
Note that you can also access a web site that will compute surfaces and map different properties on them, from Barry Honig's lab. - MSMS
- molecular surface
The program MSMS calculates analytical molecular surfaces. Make sure before reading the *.face file, that the 2 result files have the correct names *.face and *.vert and are located in the same directory. [Ref: Michael F. Sanner, Olson & Spehner, Biopolymers (1996) 38, 305]