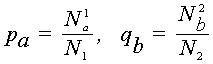
ON THE OPTIMALITY OF THE DAYHOFF MATRIX FOR COMPUTING THE SIMILARITY SCORE BETWEEN FRAGMENTS OF BIOLOGICAL SEQUENCES
A.M. Leontovich
Institute of Physico-chemical Problems in Biology, laboratory bldg. A,
Moscow State University, Moscow 119899, Russia
abstract. One of the most important problems of the biological sequence analysis is the search for similar sequences or sequence fragments and measuring the observed similarity. One of the most widespread and theoretically substantiated definitions of the similarity measure is that by Staden. The exact numerical value of this measure depends on the choice of the letter substitution weight matrix that characterizes the similarity of individual letters (nucleotides or amino acid residues) constituting the compared sequences. In the case of amino acid sequences the most often used matrix is that by Dayhoff. Its construction is based on the statistics of amino acid substitutions in a set of a priori related (but sufficiently divergent) proteins. In this paper some theoretical corroboration for the Dayhoff matrix are presented. In particular, a statement is proved showing that in some sense the Dayhoff matrix is an optimal matrix for the search of similar fragments.
In the analysis of biological sequences (polynucleotide or polypeptide) it is often necessary to understand whether there is a similarity between these sequences or their fragments, and, if such a similarity exists, to estimate how large it is. One of the most widespread and theoretically substantiated definitions of a similarity measure is that by Staden [1]. The exact numerical value of this measure depends on the choice of letter substitution weights; these weights characterize the similarity of individual letters (nucleotides or amino acid residues), that constitute the compared sequences.
The choice of a matrix can vary for different situations. The simplest matrix is the unit matrix with weights equal 1 if the letters coincide and 0 if the letters differ. The unit matrix is usually considered as the weight matrix if polynucleotide sequences are compared. But in the case of polypeptide sequences other matrices are mostly used [2, p. 103]. Sometimes the choice of weights depends on certain physical chemistry considerations (for instance, the hydrophobicity of amino acids, the charge, and so on) [3]. Another method of the weight matrix construction stems from the genetic code table [2, p. 103], [4]. Finally, a very often (probably the most often) used matrix is the Dayhoff matrix [4], whose construction is based on the statistics of letter (amino acid) substitutions (it seems that biologists do not use this approach for analysis of polynucleotide sequences). In this matrix relatively large weights correspond to letter pairs with the tendency for mutual substitution (of course, large weights correspond also to coinciding letters).
The last choice (the Dayhoff matrix) is the object of this paper. It will be shown that in some sense the Dayhoff matrix is an optimal matrix for the search of similar fragments.
First, let us recall the definition of the Staden similarity measure for fragments.
Consider two biological sequences, either polynucleotide (DNA or RNA), or polypeptide (proteins). Let a1,...,aN1 and b1,...,bN2 be the letters constituting these sequences. If the polynucleotide sequences are being considered, then each of letters ar, bs is one of the four nucleotide bases (A, G, C, T/U). If polypeptide sequences are being considered, then ar, bs belong to the set of 20 amino acids. Denote by pa , qb the frequencies of the letters a and b in the first and the second sequences respectively, that is
(1)
where N1a is the number of occurences of the letter a in the first sequence (i.e. the number of subscripts l for which al = a,
1  l
n1), N2b is the number of occurences of the letter b in the second sequence, and N1 and N2 are lengths of sequences.
l
n1), N2b is the number of occurences of the letter b in the second sequence, and N1 and N2 are lengths of sequences.
For each pair of letters a, b we introduce the number r a,b that characterizes the similarity of the letters a and b. This number is called the weight of the substitution of the letter a to the letter b. The weights pa,b form the matrix P = ||r a,b||. For polynucleotide sequences the matrix P is of dimension 4x4, while for polypeptides it is of dimension 20 x 20. Denote by m and D the mean and the variance of substitution weights in considered sequences:
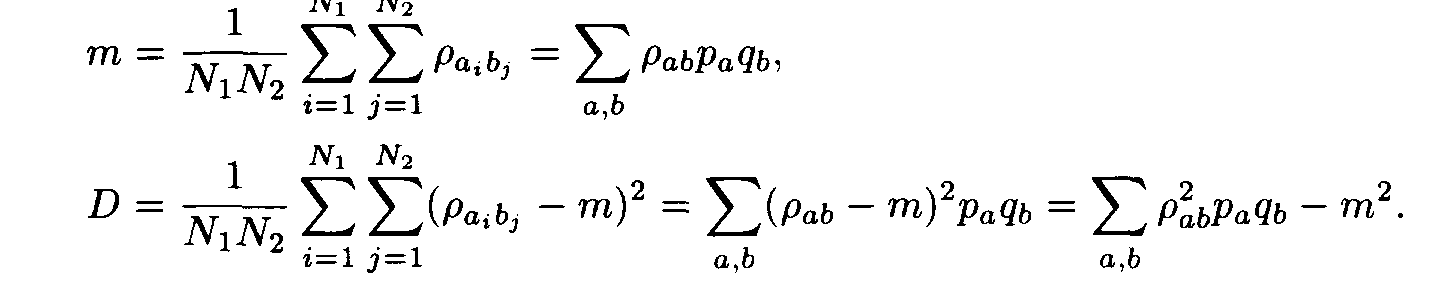
Let (ai, ai+1,..., ai+L-1) and (bj, bj+1,... ,bj+L-1) be a pair of the sequence fragments of length L. Consider
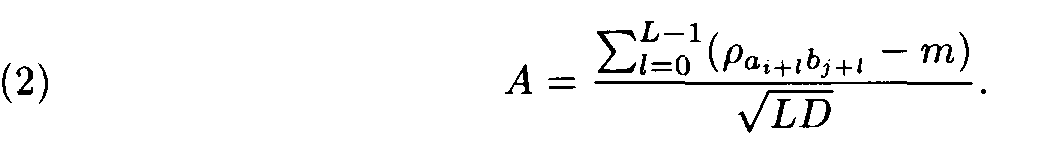
This value A was suggested by Staden as a similarity measure between the compared fragments (ai,ai+1,... ,ai+L-1) and (bj,bj+1,...,b j+L-1).
If we assume that the sequences (a1,...,aN1), (b1,...,bN2) are independent Bernoulli sequences with letter frequencies pa, qb respectively, then the weights r
a(i+l), b(j+l) are random variables with the mean m and the variance D, and these random variables are independent for various l, 0 < l < L — 1. Then the similarity score A defined by formula (2) is a random variable. Its mean equals 0 and its variance equals 1. For L —>  the distribution of this variable A tends to the standard normal distribution function N(0,1), that is,
the distribution of this variable A tends to the standard normal distribution function N(0,1), that is,
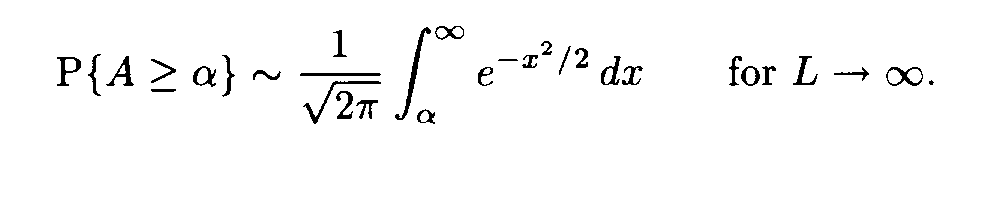
For large L this formula allows us to find the probability P of the event that the sum of substitution weights for independent Bernoulli sequences of the length L is not less than the value of the corresponding sum for the fragments under comparison. This probability P is uniquely determined by A, i.e. P = P(A). Hence A is a natural similarity measure for fragments.
Now we describe the method to construct the Dayhoff matrix (as the author understands it).
A set of protein families is considered. Each family consists of presumably related proteins. In each case the presumption of relatedness is based not only on the similarity of the corresponding sequences, but rather on the biological considerations. However, the similarity between the proteins of a given family is certain but not too large, so that the sequences are not nearly identical. On the other hand, the proteins are similar enough in order to allow an unique alignment, in which the homologous fragments of the sequences are situated below each other. Such alignment is based not only on the sequences themselves, but also on the biological and chemical properties of the proteins.
Consider a pair of proteins a ,b belonging to one family. Align the protein sequences. After the alignment lengths of the sequences become equal. Denote by N the number of the alignment positions occupied by two amino acids (and not by an amino acid and a gap). Next, denote by Naa the number of positions that are occupied by the amino acid a in the sequence a and an arbitrary amino acid (but not a deletion) in the sequence b . Similarly, denote by Nbb the number of the positions occupied by b in b and any amino acid in a . Finally, denote by Naba b the number of positions occupied by a in a and b in b . Consider
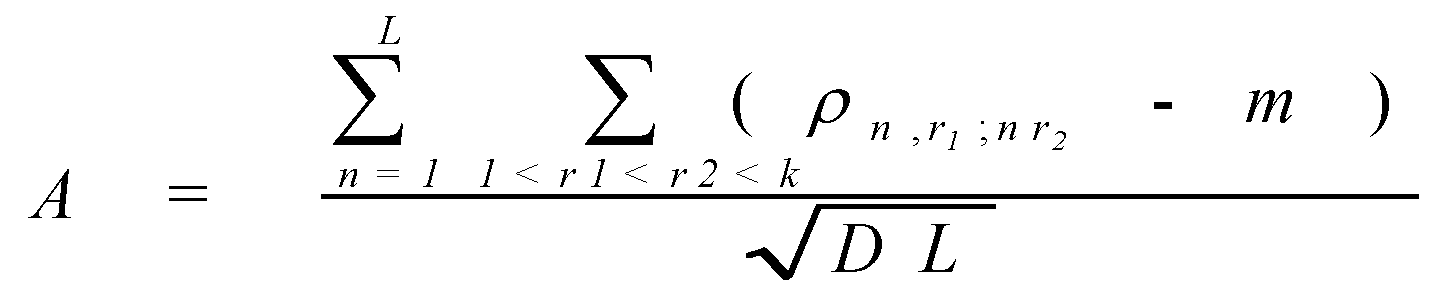
where a, b is an arbitrary pair of amino acids. (If r aba b > 1, than there is an observable tendency of the amino acid b to occupy the same position as the amino acid a). For fixed a, b the values r aba b are averaged for all protein pairs a , b belonging to same family, and then averaged for all families from the set. The obtained averaged values are considered as the weights r àb in the Dayhoff matrix. (Actually, in the real Dayhoff matrix all values are then multiplied by 10 and rounded to integers, but this is not important, since, by (2), the procedure of simultaneous multiplication of all weights by a same number does not change the value A).
Note that despite the fact that r aba b and r baa b are, in general, different, the matrix P obtained after the averaging is symmetric (that is, r ab = r ba), since the averaging is made over all protein pairs and both the pair (a , b ) and the pair (b ,a ) are considered.
The Dayhoff matrix often used by biologists is presented in Table 1 (this matrix is taken from [6, Table 1.2]).
In the Dayhoff matrix, large values of weights r ab correspond to pairs of amino acids a, b that have a tendency for mutual substitution (in particular, diagonal elements r aa have large values). This seems to be perfectly natural. But is there a more formal foundation for the Dayhoff method and formula (3). Here we present a statement that, in our opinion, partially validates this formula.
Table 1.
The Dayhoff weight matrix
|
|
Gly |
Pro |
Asp |
Glu |
Ala |
Asn |
Gin |
Ser |
Thr |
Lys |
Arg |
His |
Val |
He |
Met |
Cys |
Leu |
Phe |
Tyr |
Trp |
|
Gly |
29 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Pro |
12 |
14 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Asp |
11 |
10 |
23 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Glu |
11 |
11 |
19 |
20 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Ala |
13 |
14 |
12 |
12 |
14 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
`Asn |
11 |
10 |
14 |
12 |
12 |
17 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Gin |
10 |
11 |
14 |
14 |
11 |
12 |
21 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Ser |
12 |
11 |
12 |
11 |
13 |
13 |
11 |
13 |
|
|
|
|
|
|
|
|
|
|
|
|
|
Thr |
10 |
10 |
10 |
10 |
12 |
12 |
11 |
13 |
18 |
|
|
|
|
|
|
|
|
|
|
|
|
Lys |
7 |
8 |
10 |
10 |
9 |
12 |
11 |
10 |
9 |
24 |
|
|
|
|
|
|
|
|
|
|
|
Arg |
4 |
4 |
7 |
7 |
5 |
10 |
13 |
7 |
6 |
22 |
75 |
|
|
|
|
|
|
|
|
|
|
His |
6 |
6 |
9 |
8 |
8 |
13 |
12 |
9 |
9 |
11 |
20 |
59 |
|
|
|
|
|
|
|
|
|
Val |
7 |
8 |
7 |
8 |
10 |
8 |
9 |
9 |
11 |
8 |
5 |
6 |
22 |
|
|
|
|
|
|
|
|
He |
6 |
6 |
6 |
7 |
8 |
7 |
8 |
8 |
10 |
7 |
5 |
7 |
21 |
25 |
|
|
|
|
|
|
|
Met |
5 |
6 |
6 |
6 |
8 |
7 |
8 |
8 |
9 |
8 |
10 |
6 |
16 |
16 |
26 |
|
|
|
|
|
|
Cys |
6 |
4 |
5 |
5 |
7 |
6 |
5 |
11 |
9 |
4 |
2 |
3 |
11 |
9 |
12 |
166 |
|
|
|
|
|
Leu |
4 |
4 |
5 |
6 |
6 |
5 |
6 |
6 |
7 |
6 |
4 |
6 |
15 |
18 |
21 |
5 |
40 |
|
|
|
|
Phe |
2 |
2 |
2 |
3 |
4 |
3 |
3 |
4 |
5 |
3 |
4 |
7 |
7 |
11 |
11 |
2 |
12 |
70 |
|
|
|
Tyr |
1 |
1 |
1 |
1 |
2 |
2 |
2 |
3 |
2 |
3 |
3 |
7 |
3 |
6 |
6 |
1 |
6 |
66 |
137 |
|
|
Trp |
1 |
1 |
1 |
1 |
1 |
2 |
2 |
2 |
2 |
2 |
3 |
15 |
2 |
4 |
4 |
1 |
3 |
41 |
46 |
414 |
Let us have a pair of fragments (a1,...,aL), (b1,...,bL) of the same length L. Denote by n1a the number of positions in the first fragment occupied by the letter a, by n2b the number of positions in the second fragment occupied by the letter b, and by nàb the number of position occupied by a in the first fragment and b in the second fragment (i.e. the number of positions l such that al= a, bl = b). Following (2), the similarity measure for the two fragments is
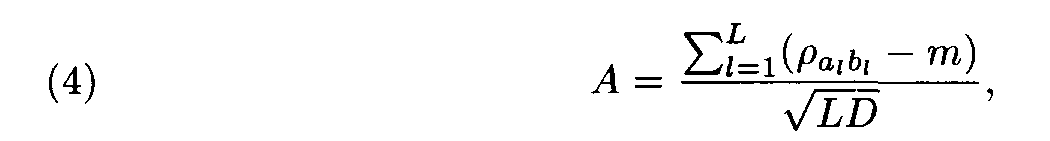
where
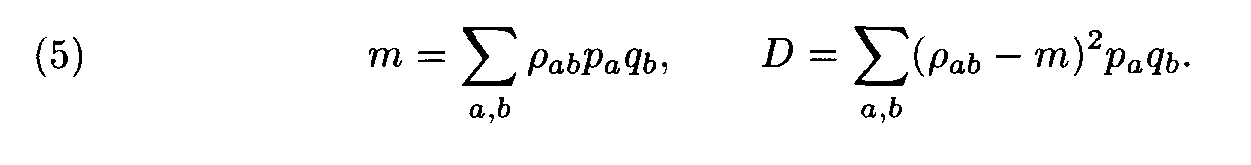
Theorem. Let

Then for each weight matrix ||r
àb|| we have
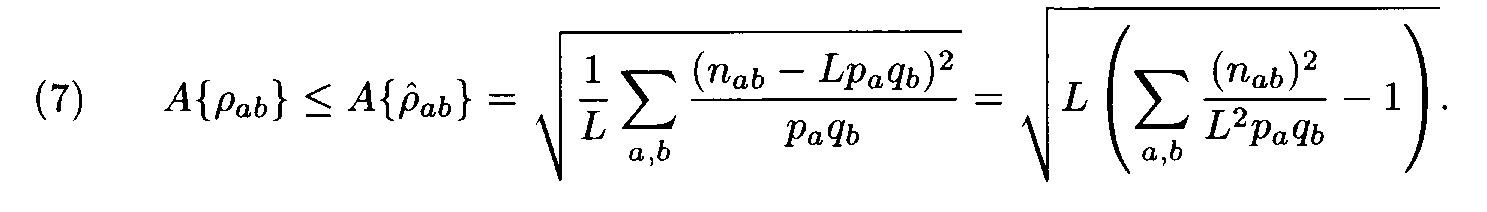
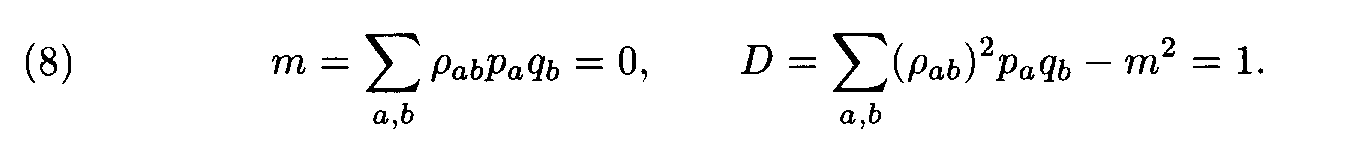
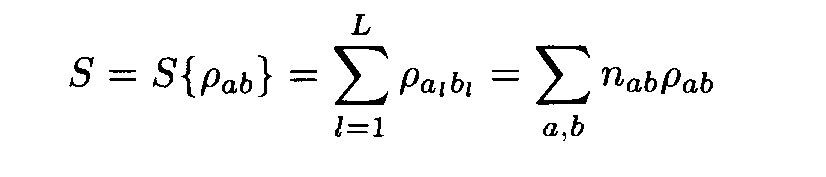
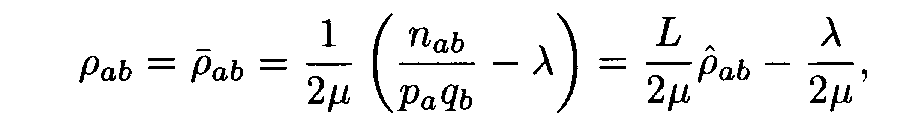
where the factors l , m are found from conditions (8). This expression implies the left inequality in (7). Next, from (5) and (6) we easily see that
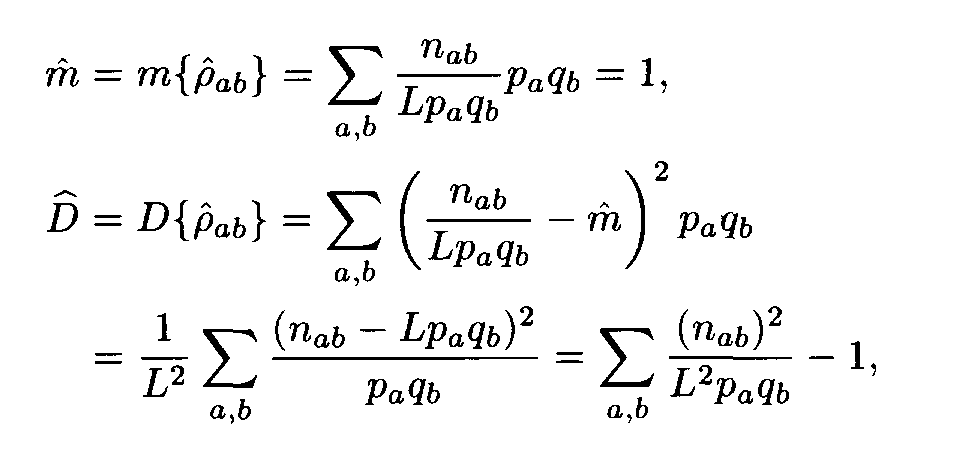
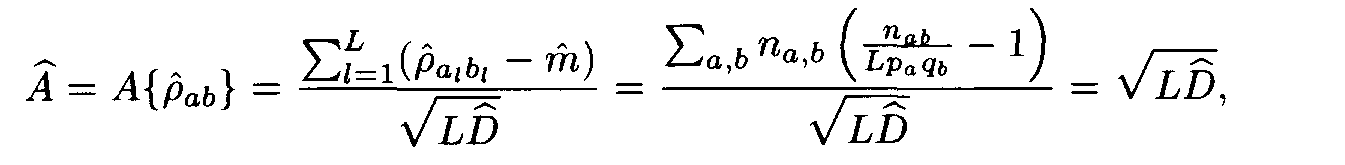
and the proof is completed.
Suppose that the letter frequencies in the fragments equal the corresponding frequencies in the entire sequences, that is
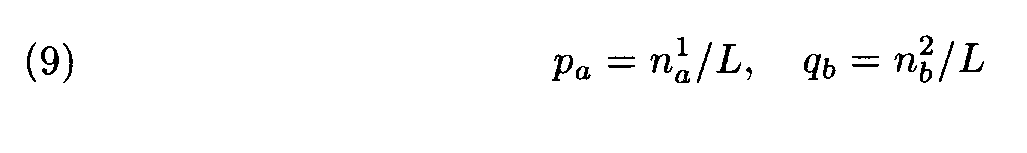
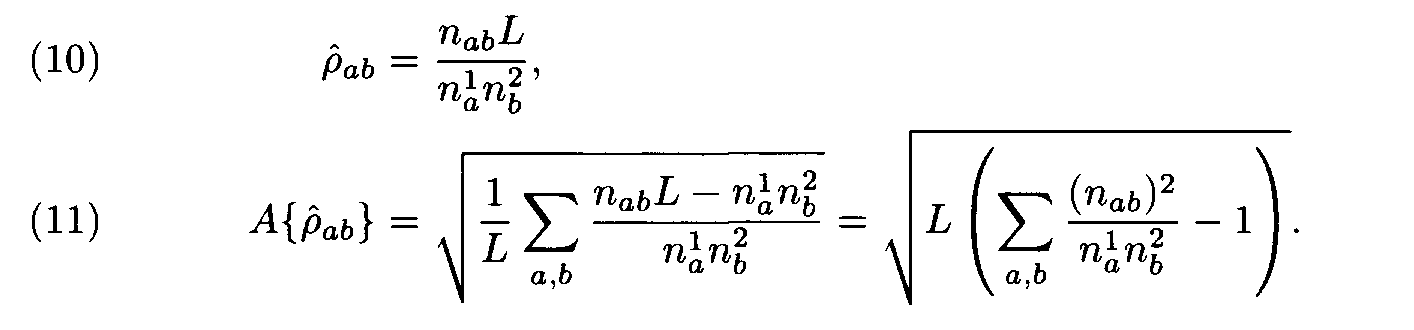
The evident similarity between formulas (3) and (10) justifies the following interpretation of the above theorem: the choice of weights for the letter (amino acid) substitutions with the Dayhoff matrix is close to optimal if the compared sequences (proteins) have the intermediate similarity that is close to the similarity level in the protein families used as learning sets in the construction of the Dayhoff matrix. If the similarity level is different, some other matrix should be used.
As an example we consider the case of nearly identical sequences. For such sequences the values na1, na2, naa are close to each other, while the values ïàb are small compared to na1, na2 for à a ¹ b. Formula (10) implies that in this case it is useful to choose as a weight matrix the diagonal matrix with elements inversely proportional to letter frequencies. (However, for such almost coinciding sequences the choice of the weight matrix is of no great importance since similar fragments would be surely found with the unit matrix, Dayhoff matrix, or many other matrices.)
Formulas (6) and (10) suggest possible modifications of the Staden approach to the definition of the similarity measure for fragments. In these modifications for the letter substitutions are not set once and forever, but depend of the sequences being considered. Two approaches are possible.
In the first approach the recomputation of weights is made during process of determination of similarity between fragments. This recomputation of weights depends on the sequences as a whole, but not on the fragments under consideration. First, some standard weight matrix ||r àb|| (e.g., the Dayhoff matrix) is considered and similar fragments are found. Then the alignment is performed. Hence, the similarity level of those two sequences is determined. If necessary, weights are redefined using the following procedure. Denote by N the number of the alignment positions occupied by letters in both sequences (and not a letter and a gap). Denote by Na1 the number of positions occupied by the letter a in the first sequence and by an arbitrary letter in the second sequence; similarly, denote by n b 2 the number of the positions occupied by by the letter b in the second sequence. Finally, denote by Nab the number of the positions occupied by a in the first sequence and by b in the second sequence. The new weights are then defined by the formula
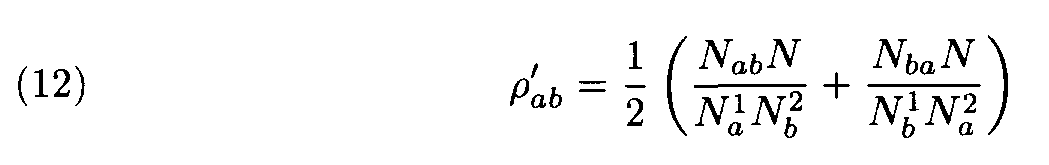
(cf. (3), (10); note that the matrix ||r
’ àb|| is symmetric since for that very reason its elements in (12) are defined as half sums of the two terms). Then the more accurate search of similar fragment is performed using these new more adequate weights
r
'ab.
When this procedure is applied to independent Bernoulli sequences, the distribution of the random variable A' characterizing the similarity between fragments differs from the distribution of the corresponding random variable A defined using the preset weight matrix r
àb. In particular, it is evident that the distribution of A' is shifted to the right compared to the distribution for A, and that although A' has an asymptotically normal distribution N(0, 1) for L —>
, N / L —>
, it tends to the limit distribution more slowly than A. These facts should be considered during interpretation of observed results.
The described approach has an evident drawback compared to the standard Staden method. It is too complicated. Indeed, in order to find the weights ||r 'ab||, the usual Staden procedure is to be performed and then the sequences are to be aligned, which is a laborous task.
In the second approach the weights depend directly on the fragments that are compared, and not on the entire sequences from which the fragments were taken. Namely, it is suggested to use as the weights the values r àb maximizing the similarity measure between the fragments (see (6)). Then the similarity measure between the fragments Af is defined by formula (7), so that Af =
A{r fàb}. In this case letter frequencies (that are parts of (6) and (7)) can be computed using either the entire sequences (see (1)) or the fragments under consideration. (See (9). The weights r fàb and the similarity measure Af = À{r f àb} are defined by formulas (10) and (11).) It is more convenient to compute not the similarity measure Af, but its square Af 2. In this approach the recomputation of the weights is not performed, so it is less cumbersome. Moreover, the simplicity of formulas (7), (11) also is attractive.
To interpret the experimental results it is necessary to understand the nature of the distribution of the variable Af (or Af 2) when independent Bernoulli sequences are compared. Evidently, the distribution of Af is fundamentally different from the distributions of A and A' discussed above. For example, the value Af (unlike A and A') is automatically non-negative (see (7)). Of course, the distribution of Af for L —>
does not tend to the standard normal distribution N(0, 1).
Consider in more detail the case when the letter frequencies are computed using the entire sequences (i.e. by formula (1)). We analyse the distribution of Af 2. By (7), Af 2 is the sum of 202 = 400 positive terms

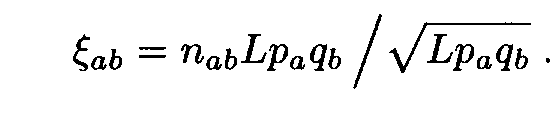
It is not difficult to compute
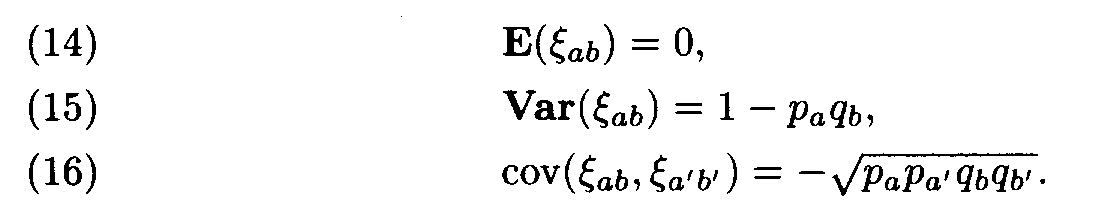
It can be shown that the variables x
ab, are asymptotically normal for L —>
. (Therefore, Af 2 looks like the x2 with 400 degrees of freedom, but by (15), the variances of x
ab differ from 1, and, what is even more important, by (16) these values are mutually dependent.) Formulas (14) and (15) imply that
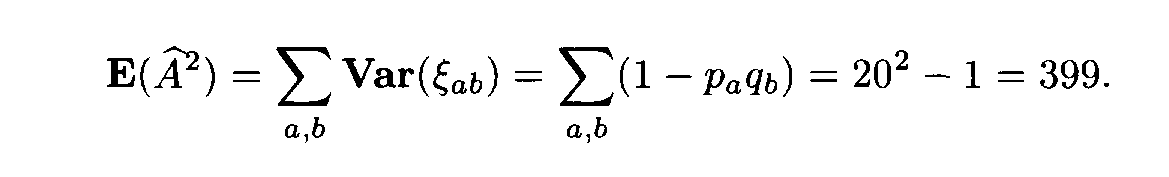
In the case when all letters have equal frequencies pa = qb = 1/20, it is possible to find also the limit value (for L —>
) for the variance Var(Af 2). Namely, computing the eigenvalues of the covariation matrix (15), (16), one sees that one of the eigenvalues equals 0, while all others equal 1. Using the asymptotic normality of the variables x
ab, it is possible to prove that in this case
Var(Af 2) —> 2(202 - 1) = 798 for L —>
.
In the general case we cannot compute the variance Var(Af 2), since the eigenvalues of the covariation matrix cannot be found. However, the limit value of the variance would be certainly close to 798, even if the letter frequencies are not close to one another.
The variable Af 2 is not, of course, asymptotically normal for L —>
(as mentioned above, it is more close to the variable x2 with 399 degrees of freedom). However, since the number of terms in (13) is very large, the distribution of Af 2 is close to normal. Therefore in applications one can assume that Af 2 has a normal distribution with the mean 399 and the variance 798.
Since the mean E(Af 2) is very large, the distribution of the variable Af also is close to normal with the mean close to
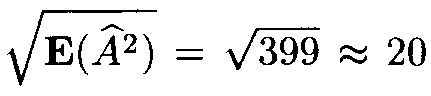
and the variance close to
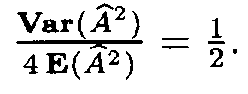
A similar situation for the distribution of (Af 2 and (Af occures in the case when the letter frequencies are computed from the fragments, i.e. by formula (9). However, in this case the mean Å((Af 2) and the variance Var((Af 2) are slightly different:
E((Af 2) =
202 - 2 • 20 + 1 = 361, Var((Af 2) ~ 2 • 193/21 ~ 653(in the case of equal letter frequencies the variance coincides with the above value). As we have mentioned earlier, this approach has some advantages compared to the previous one (when the weights depend on the entire sequences and not on the compared fragments). But it has also serious drawbacks. One of them is that the value of (Af 2 is large not only if the fragments under comparison are very similar, but also when they are very dissimilar (i.e. when there is a tendency for non-similar letters to occupy same positions). Therefore, after computing the value (Af 2 it is necessary to find out why this value is large (due to the similarity or the dissimilarity of the fragments). The other drawback is that this approach (contrary to both the standard Staden technique and the first alternative technique) cannot be used by the DotHelix algorithm for the fast search of similar fragments in sequences [5].
Hence, both methods have serious drawbacks (besides the fact that they have not been studied analytically in any detail). On the other hand, the problem of the weight choice seems to be not too important for biologists. Nevertheless, the possibility of such approaches is worth bearing in mind.
References
[1] R. Staden, An interactive graphics program for comparing and aligning nucleic acid and ammo acid sequences, Nucl. Acids Res. 10 (1982), 2951-2961.
[2] R. F. Doolittle, Of URFs and ORFs. A primer on how to analyze derived amino acid sequences, University Science Books, Mill Valley, 1986.
[3] W. M. Fitch, An improved method of testing for evolutionary homology, J. Mol. Biol. 16 (1966), 9-16.
[4] M. D. Dayhoff, Atlas of protein sequence and structure, vol. 5, suppl. 3, Natl. Biomed. Res. Found., Washington, D.C., 1978.
[5] A. M. Leontovich, L. I. Brodsky, and A. E. Gorbalenya, Construction of the complete map of local similarity for two polymers, Biopolimery i Kletka 6 (1990), no. 6, 14-21. (Russian)
[6] G. E. Shulz and R. H. Schirmer, Principles of protein structure, Springer-Verlag, New York, 1979.